

How can we revolutionize CO2 capture? Computational design to the rescue of the climate
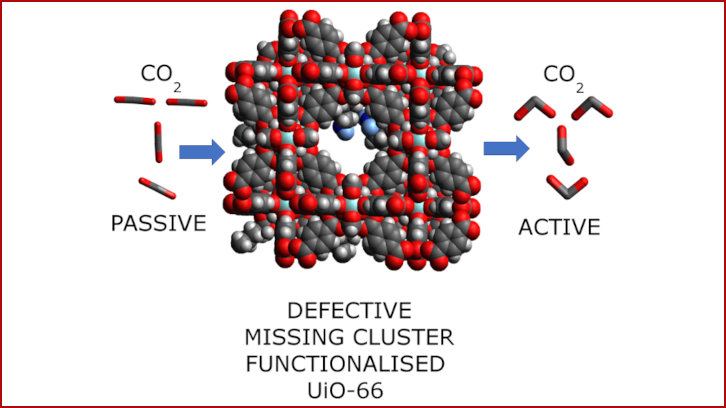
Capturing carbon dioxide (CO2) is one of the most promising strategies for mitigating emissions. For this reason, the Department of Chemistry aims to revolutionize the way it is captured. With the help of computational design, they have presented new strategies and advances in the optimization of MOFs, materials with sponge-like pores that can soak up gases.
The fight against climate change is urgent, and capturing carbon dioxide (CO2) is one of the most promising ways to make a difference. In our latest research, we set out to design materials that could revolutionize how we trap and store CO2. Our focus? Metal-organic frameworks (MOFs), remarkable materials with sponge-like pores that can soak up gases. But we didn’t stop there. We wanted to make these MOFs even smarter by giving them a chemical edge.
Using a popular MOF called UiO-66, we introduced amine groups, molecular "hooks" that can grab onto CO2 molecules. By grafting amino acids onto the internal surfaces of the MOF, we explored how chain length affects their CO2-trapping abilities. Through computational experiments, we found that not all amines are created equal. Short-chain amino acids like glycine and beta-alanine just didn’t get close enough to work together. But when we used longer chains, like gamma-aminobutyric acid and 5-aminovaleric acid, something remarkable happened. The longer chains allowed the amine groups to form hydrogen bonds, stabilizing CO2 molecules inside the MOF.
This cooperative interaction even triggered a fascinating transformation, turning CO2 into carbamic acid through a double hydrogen transfer. What’s even more exciting is how efficient these modifications can be. For example, with 5-aminovaleric acid, functionalizing just 16% of the available sites was enough to capture CO2 effectively. That’s a big win for making these materials cost-efficient and scalable. We also found that tweaking the amines by adding protons could supercharge their CO2 interactions, offering another way to optimize these systems.
Our work doesn’t stop here. We’re already in the lab, synthesizing and testing these modified MOFs to bring our designs to life. By combining computational insights with experimental validation, we’re paving the way for next-generation materials that could play a key role in reducing CO2 emissions. It’s not just about capturing carbon, it’s about creating a greener, more sustainable future. And we’re excited to be part of that journey.
Gerard Pareras Niell; Albert Rimola Gibert
Department of Chemistry
Universitat Autònoma de Barcelona
Marco Taddei
Department of Chemistry and Industrial Chemistry
Università di Pisa
Davide Tiana
School of Chemistry
University College Cork
References
Pareras, G., Rimola, A., Taddei, M., Tiana, D. (2024). Computationally aided design of defect-appended aliphatic amines for CO2 activation within UiO-66. Physical Chemistry Chemical Physics, 26(42), 26958-26965. https://doi.org/10.1039/D4CP03223C
