¿Cómo podemos revolucionar la captura del CO2? Diseño computacional al rescate del clima
Capturar dióxido de carbono (CO2) es una de las estrategias más prometedoras para mitigar las emisiones. Por eso, desde el Departamento de Química se proponen revolucionar la manera en que se atrapa. De la mano del diseño computacional, han presentado nuevas estrategias y avances en la optimización de los MOFs, materiales con poros similares a esponjas que pueden absorber gases.
La lucha contra el cambio climático es urgente, y capturar dióxido de carbono (CO2) es una de las estrategias más prometedoras para marcar la diferencia. En nuestra investigación, nos propusimos diseñar materiales capaces de revolucionar la manera en que atrapamos y almacenamos CO2. ¿Nuestro enfoque? Los metal-organic frameworks (MOFs), materiales fascinantes con poros similares a esponjas que pueden absorber gases. Pero no nos detuvimos ahí: quisimos hacer que estos MOFs fueran aún más versátiles añadiendo modificaciones posteriores.
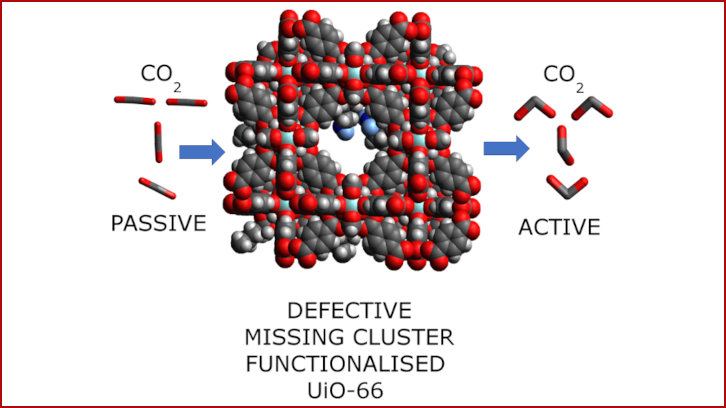
Utilizando el MOF “UiO-66”, incorporamos grupos amino que actúan como “ganchos” moleculares para atrapar las moléculas de CO2. Al insertar aminoácidos en las superficies internas del MOF, analizamos cómo la longitud de la cadena afecta su capacidad para capturar CO2. Gracias a experimentos computacionales, descubrimos que no todas las aminas funcionan de la misma manera. Los aminoácidos de cadena corta, como la glicina y la beta-alanina, no estaban lo suficientemente cerca entre sí para cooperar. Sin embargo, al emplear cadenas más largas, como el ácido gamma-aminobutírico y el ácido 5-aminovalérico, ocurrió algo extraordinario. Estas cadenas más largas permitieron que los grupos amino formaran enlaces de hidrógeno, estabilizando las moléculas de CO2 dentro del MOF.
Esta interacción cooperativa incluso dio lugar a una fascinante transformación: la conversión del CO2 en ácido carbámico mediante un mecanismo de transferencia doble de hidrógeno. Lo más emocionante es la eficiencia de estas modificaciones. Por ejemplo, en el caso del ácido 5-aminovalérico, bastó con modificar solo el 16% de los sitios disponibles para capturar CO2 de manera efectiva. Esto supone un avance significativo hacia el desarrollo de materiales más económicos y escalables. También descubrimos que la protonación de las aminas puede potenciar aún más su interacción con el CO2, ofreciendo otra vía prometedora de optimización.
Nuestro trabajo no termina aquí. Ya estamos en el laboratorio, sintetizando y probando estos MOFs modificados para llevar nuestros diseños a la realidad. Combinando perspectivas computacionales con validación experimental, estamos allanando el camino hacia materiales de nueva generación que podrían desempeñar un papel clave en la reducción de las emisiones de CO2. Esto no se trata solo de capturar carbono, sino de construir un futuro más verde y sostenible.
Gerard Pareras Niell; Albert Rimola Gibert
Departamento de Química
Universitat Autònoma de Barcelona
Marco Taddei
Departamento de Química y Química Industrial
Università di Pisa
Davide Tiana
School of Chemistry
University College Cork
Referencias
Pareras, G., Rimola, A., Taddei, M., Tiana, D. (2024). Computationally aided design of defect-appended aliphatic amines for CO2 activation within UiO-66. Physical Chemistry Chemical Physics, 26(42), 26958-26965. https://doi.org/10.1039/D4CP03223C